Cyclic Peptide Structure Prediction via Structural Ensembles Achieved by Molecular Dynamics and Machine Learning (StrEAMM)
Case ID: T002519
Web Published: 4/11/2022
Description:
Efficient Development of Orally-Bioavailable Cyclic-Peptide Drugs via Molecular Dynamics + Machine Learning
BACKGROUND
Cyclic peptides are a promising drug modality, combining beneficial traits from both small molecules and antibody or protein therapeutics. While many scientists think that cyclic peptides are key to providing orally-available therapeutics, there are only a few FDA-approved cyclic peptides due to the complexities of cyclic peptide drug discovery.
PROBLEM
One of the main challenges in developing efficient cyclic peptides is due to their chameleonic properties as they tend to adopt multiple confirmations in a solution, which are difficult to characterize and predict.
solution
Dr. Yu-Shan Lin’s team at Tufts University has combined molecular dynamic simulations to train machine learning models to efficiently predict cyclic peptide confirmation. This method is called StrEAMM (Structural Ensembles Achieved by Molecular Dynamics and Machine Learning). StrEAMM is the first technique capable of efficiently predicting complete structural ensembles of cyclic peptides without relying on additional molecular dynamics simulations, constituting a seven-order-of-magnitude improvement in speed while retaining the same accuracy as explicit-solvent simulations. Furthermore, the team is actively working to use structural data to train a predictive model that connects the confirmation to desirable drug properties like binding affinity and membrane permeability.
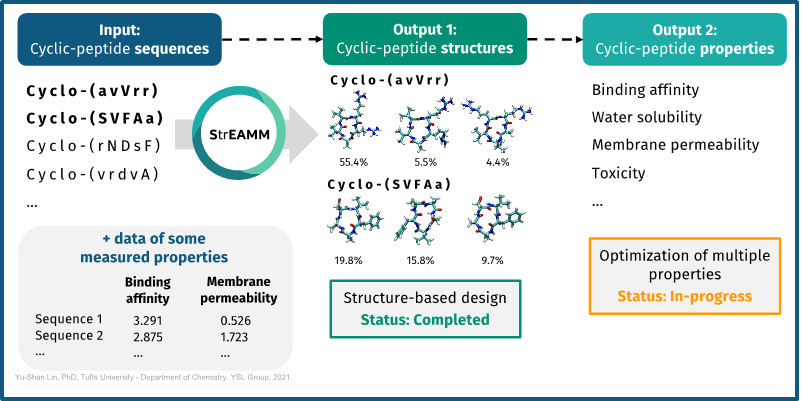
Advantages
- Distills experimental data to provide a predictive model (ML) and suggest next generation designs
- Enables rational design of cyclic peptides with desired structures and favorable drug properties
- The prediction for each individual cyclic peptide can be made in less than 1 second of computation time, dramatically reducing cycle time
Publication: Structure prediction of cyclic peptides by molecular dynamics + machine learning (November, 2021).
IP Status: Two provisional applications filed (6/14/2021 & 10/14/2021)
Patent Information:
- App Type
- Country
- Serial No.
- Patent No.
- File Date
- Issued Date
- Expire Date
Direct Link: http://tufts.technologypublisher.com/technology/46396
Category(s):
Screening: combinatorial chemistry
Software
Share
Inventors:
Keywords:

